一、目的
明确药物临床试验中安全性事件报告的流程。
二、适用范围
适用于本机构所有药物临床试验。
三、标准操作规程
1 安全性事件包含了严重不良事件、可疑且非预期严重不良反应和研发期间安全性更新报告。
1.1 严重不良事件(Serious Adverse Event,SAE):受试者接受试验用药品后出现死亡、危及生命、永久或者严重的残疾或者功能丧失、受试者需要住院治疗或者延长住院时间,以及先天性异常或者出生缺陷等不良医学事件。
1.2 可疑且非预期严重不良反应(Suspected and Unexpected Serious Adverse Reactions,SUSAR):临床表现的性质和严重程度超出了试验药物研究者手册、已上市药品的说明书或者产品特性摘要等已有资料信息的可疑并且非预期的严重不良反应。
1.3 研发期间安全性更新报告(Development Safety Update Report,DSUR),是申办者对报告周期内收集到的与在研药物(无论上市与否)相关的安全性信息进行全面深入的年度回顾和评估。
2 上报要求
2.1 SAE的上报:除试验方案或者其他文件(如研究者手册)中规定不需立即报告的严重不良事件,研究者应当立即向申办者书面报告所有严重不良事件,随后应当及时提供详尽、书面的随访报告。严重不良事件报告(见附件1)和随访报告应当注明受试者在临床试验中的鉴认代码,而不是受试者的真实姓名、公民身份号码和住址等身份信息。
2.2 试验方案中规定的、对安全性评价重要的不良事件和实验室异常值,应当按照试验方案的要求和时限向申办者报告。
2.3 涉及死亡事件的报告,研究者应当向申办者和伦理委员会提供其他所需要的资料,如尸检报告和最终医学报告。
2.4 研究者收到申办者提供的临床试验的相关安全性信息后应当及时签收阅读,并考虑受试者的治疗,是否进行相应调整,必要时尽早与受试者沟通,并应当向伦理委员会报告由申办方提供的可疑且非预期严重不良反应。
2.5 申办者提供的药物研发期间安全性更新报告应当包括临床试验风险与获益的评估,有关信息通报给所有参加临床试验的研究者(需审阅签字)及临床试验机构、伦理委员会。
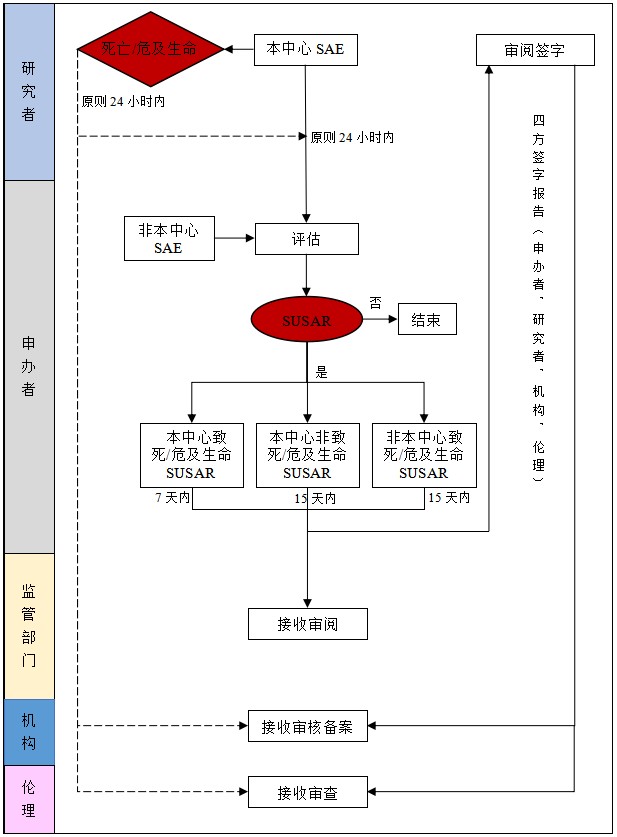
3 上报流程
3.1 在药物临床试验进行过程中受试者发生SAE(非试验方案或者其他文件中规定不需立即报告的严重不良事件),研究者立即(原则上24小时内)上报申办者,申办者应当立即分析评估,包括严重性、与试验药物的相关性以及是否为预期事件等。致死或危及生命的SUSAR,申办者在获知后首次7天内上报,并在随后的8天内报告、完善随访信息;本中心发生的非致死或危及生命的SUSAR或其他临床试验中心发生的致死或危及生命的SUSAR,申办者在首次获知后15天内报告。申办者获知的当天为第0天。同时申办者应当向药品监督管理部门和卫生健康主管部门报告SUSAR。SUSAR报告由申办者签字后报送研究者,研究者审阅签字后报送机构办公室和伦理,共同签字确认。其他中心发生的非致死或危及生命的SUSAR则以按照监查频率递交报告。
3.2 申办者和研究者在非预期且严重的不良事件与药物因果关系判断中不能达成一致时,其中任何一方判断不能排除与试验药物相关的,都应该进行快速报告。申办者应将SUSAR快速报告至所有参加临床试验的研究者及临床试验机构、伦理委员会;药品监督管理部门和卫生健康主管部门。注:以下情况一般不作为快速报告内容:①非严重不良事件;②严重不良事件与试验药物无关;③严重但属预期的不良反应;④当以严重不良事件为主要疗效终点时,不建议申请人以个例安全性报告(ICSR),建议在方案中写清楚。
3.3 SAE中,引起受试者死亡、危及生命的情形应当予以特别关注,因为此类事件的发生都并非开展临床试验所期望的结局,因此无论是否最后判定为SUSAR,如试验机构和伦理委员会认为有必要及时获知并采取措施控制试验风险时,可根据承接试验风险和本机构实际情况等,增加此报告要求(参见附件2虚线部分)。
4 报告方式
将《严重不良事件报告表》(原件扫描)以“项目名称+受试者筛选号+不良反应名称+报告类型”为标题发送至伦理邮箱(lhyygcplunli@163.com)和机构办公室邮箱(lhyy_ctc2018@163.com)。后续及时递交纸质版文件至机构和伦理。
严重不良事件报告流程图
 微信服务号
微信服务号 微信订阅号
微信订阅号