一、目的
明确医疗器械(包含体外诊断试剂,下同)临床试验严重不良事件报告的标准操作规程。
二、适用范围
适用于本机构所有医疗器械临床试验。
三、标准操作规程
1 严重不良事件(Serious Adverse Event,SAE),是指医疗器械临床试验过程中发生的导致死亡或者健康状况严重恶化,包括致命的疾病或者伤害、身体结构或者身体功能的永久性缺陷、需住院治疗或者延长住院时间、需要采取医疗措施以避免对身体结构或者身体功能造成永久性缺陷;导致胎儿窘迫、胎儿死亡或者先天性异常、先天缺损等事件。
2 处理与记录
研究者参照《不良事件和严重不良事件处理与记录的标准操作规程》有关章节,对SAE进行及时处理与记录。
3 相关性判断
主要研究者按照《不良事件和严重不良事件处理与记录的标准操作规程》有关章节,对SAE与试验用医疗器械的相关性进行判断,必要时可邀请有关专家参与。
4 报告流程
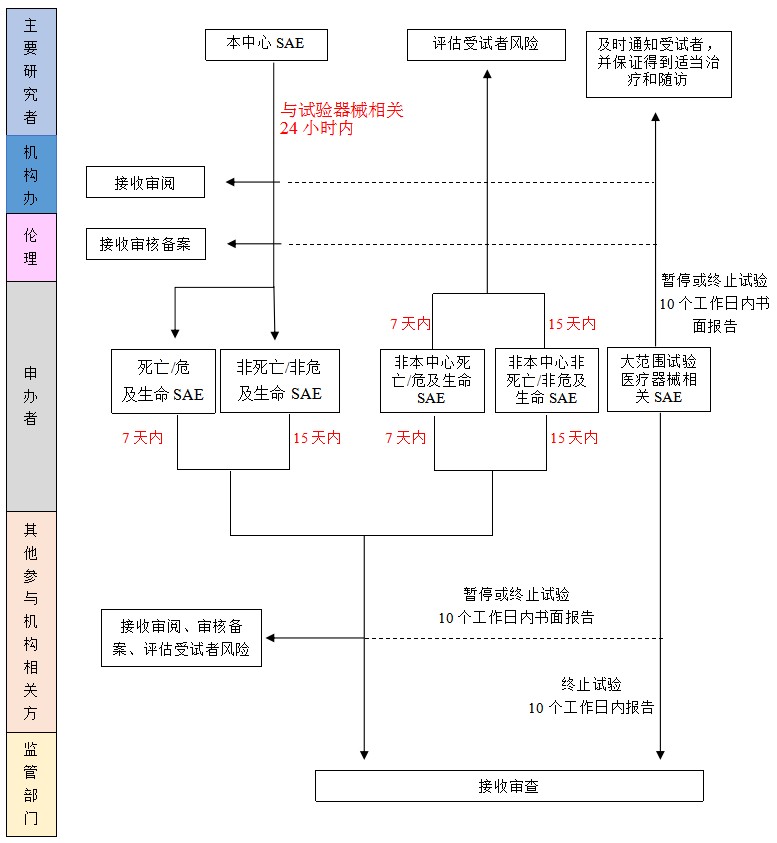
4.1报告时限:研究者在获知受试者出现SAE时,应按方案、项目标准操作规程立即采取适当的治疗措施,并在24小时内报告申办者、临床试验机构办公室(电话0755-82182992)、伦理委员会,并按照临床试验方案的规定进行随访,提交SAE随访报告。申办者应当在获知死亡或者危及生命的临床试验医疗器械相关SAE后7日内、获知非死亡或者非危及生命的试验医疗器械相关SAE后15日内,向参与临床试验的其他医疗器械临床试验机构、伦理委员会以及主要研究者报告,并向申办者所在地省(自治区、直辖市)药品监管部门、广东省药品监督管理部门和卫生健康管理部门报告,并采取风险控制措施。
4.2 出现大范围临床试验医疗器械相关SAE或者其他重大安全性问题时,申办者应当暂停或者终止医疗器械临床试验,并向所有医疗器械临床试验机构、伦理委员会以及主要研究者报告,向申办者所在地省(自治区、直辖市)药品监督管理部门、所有医疗器械临床试验机构所在地省(自治区、直辖市)药品监督管理部门和卫生健康管理部门报告。
4.3 主要研究者在收到申办方提供的试验医疗器械相关SAE和其他安全性信息时,应当及时签收阅读,并考虑受试者的治疗是否进行相应调整,必要时尽早与受试者沟通。
4.4 广东省药品监督管理局报告方式
邮件收件地址:广东省广州市越秀区东风东路753-2号 广东省药品监督管理局;收件人:医疗器械监管处;电话:37885525;传真:37885407。
4.5广东省卫生健康委员会报告方式
邮寄(使用EMS)收件地址:广州市越秀区先烈南路17号大院卫健委办公大楼;收件人:医政处;电话:020-83828646;传真:020-83805506。
5 追踪随访
5.1 医疗器械临床试验中发生SAE时,研究者应当立即对受试者采取适当的治疗措施,并按照临床试验方案的规定进行随访,提交SAE随访报告。
5.2 如SAE发生转归或距发生之日相隔30日,需填报随访报告或总结报告。
5.3 如判断与试验用医疗器械相关的SAE,必须随访至该事件出现转归、结束或受试者失访;如判断与试验用医疗器械不相关的SAE,必须随访至该事件发生后的第28天;如因妊娠上报SAE的,应随访至胎儿出生,观察有无致畸、致残等事件发生。
医疗器械/体外诊断试剂临床试验SAE报告流程图
 微信服务号
微信服务号 微信订阅号
微信订阅号